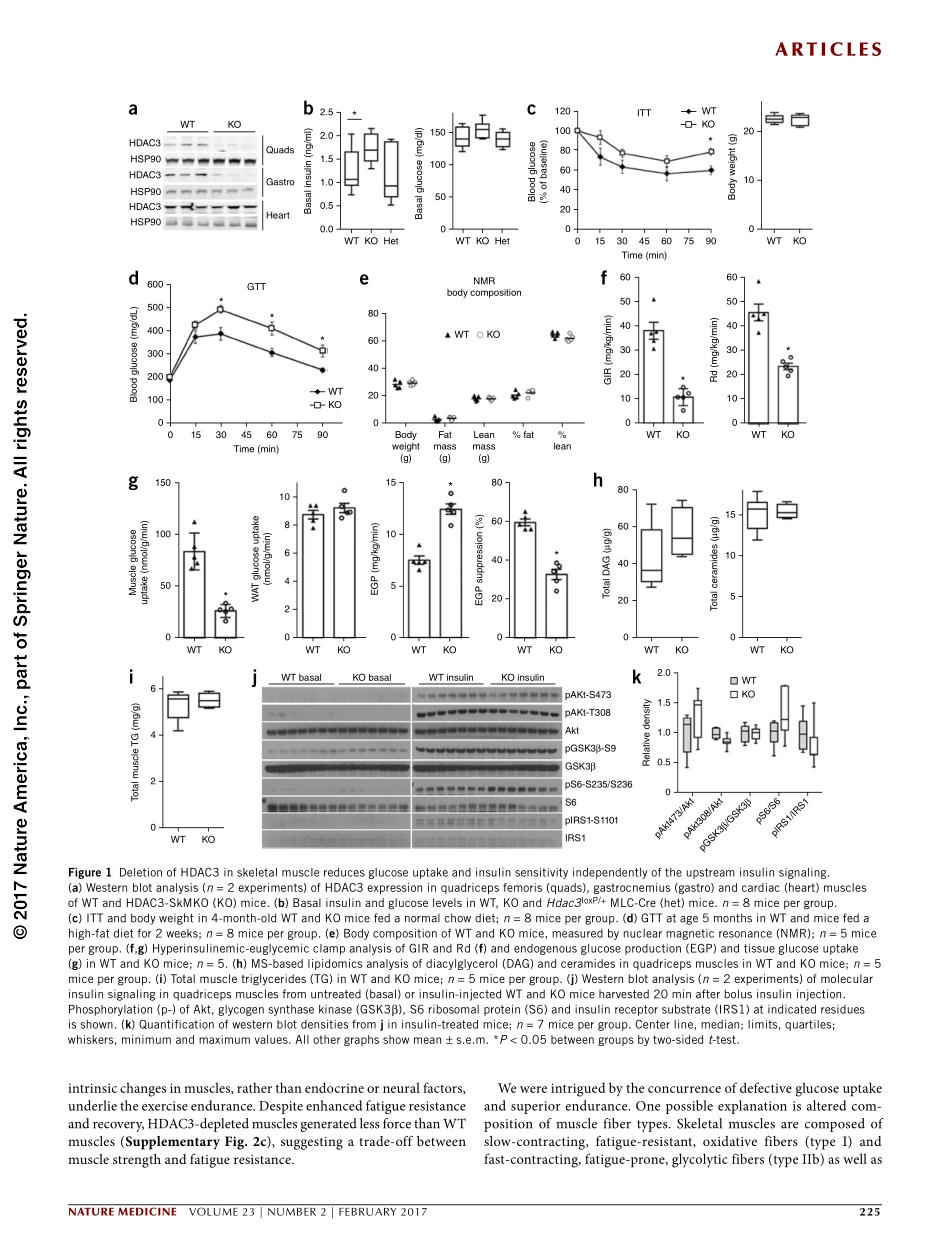
articlesnaturemedicineVOLUME23|NUMBER2|FEBRUARY2017223Skeletalmuscleisthemajortissueofglucoseconsumptionandclear-ance,and,assuch,insulinresistanceinthisorganisbelievedtobeakeymediatoroftype2diabetespathogenesis1.Ectopiclipidaccumu-lationinmusclescancauseinsulinresistancebyactivatingcytosolickinasecascadesthatdisruptmolecularinsulinsignaling,aprocessreferredtoaslipotoxicity2.However,enduranceathleteshavehigherintramuscularlipidcontentsassociatedwithhigherinsulinsensitiv-ity—aphenomenonknownasthe‘athlete’sparadox’3—suggestingthathowthemusclehandleslipidstorageismoreimportantthanthelipidcontentitself.Inaddition,muscle-specificknockoutoftheinsulinreceptordoesnotincreasebloodglucoselevelsdespitecaus-ingseveremuscleinsulinresistance4,5,suggestingthatdisruptionofcytosolicinsulinsignalinginmusclemaynotbesufficienttocausesystemicglucoseintolerance.Mitochondrialdysfunctionasacauseofmuscleinsulinresistanceisdebated.Reducedskeletalmusclemitochondrialcontentandimpairedmitochondrialoxidativefunctioncorrelatewithtype2diabetes,leadingtospeculationthatmitochondrialdysfunctionunderliesinsu-linresistance.However,depletionofgenesinvolvedinmitochondrialoxidativephosphorylation(OXPHOS)inthemuscleactuallyimprovesglucosetoleranceandinsulinsensitivity,presumablythroughadaptiveenhancementofglucoseutilization6–9.Theseresultsdonotsupportmusclemitochondrialdeficiencyastherootcauseoftype2diabetes,althoughsuchseveregeneticmitochondrialdysfunctioninanimalmodelsisdifferentfromtherelativelymild,acquiredimpairmentsinmitochondrialfunctionthatoccurinhumans.Thereciprocalcom-petitionbetweenglucoseandlipidformusclefuelsourcesisknownastheRandlecycleandprobablyinvolvesmechanismsbeyondtheallostericenzymaticregulationoriginallydefinedbyRandle10,11.Anemergingparadigmisthatmetabolicinflexibility,causedbynutri-entoverloadandheightenedfuelcompetition,preventsefficientutilizationofanyfuelandleadstoaccumulationoftoxicintermedi-atesandinsulinresistance12,13.Reducedmitochondrialactivityinthismodelisacquireda...