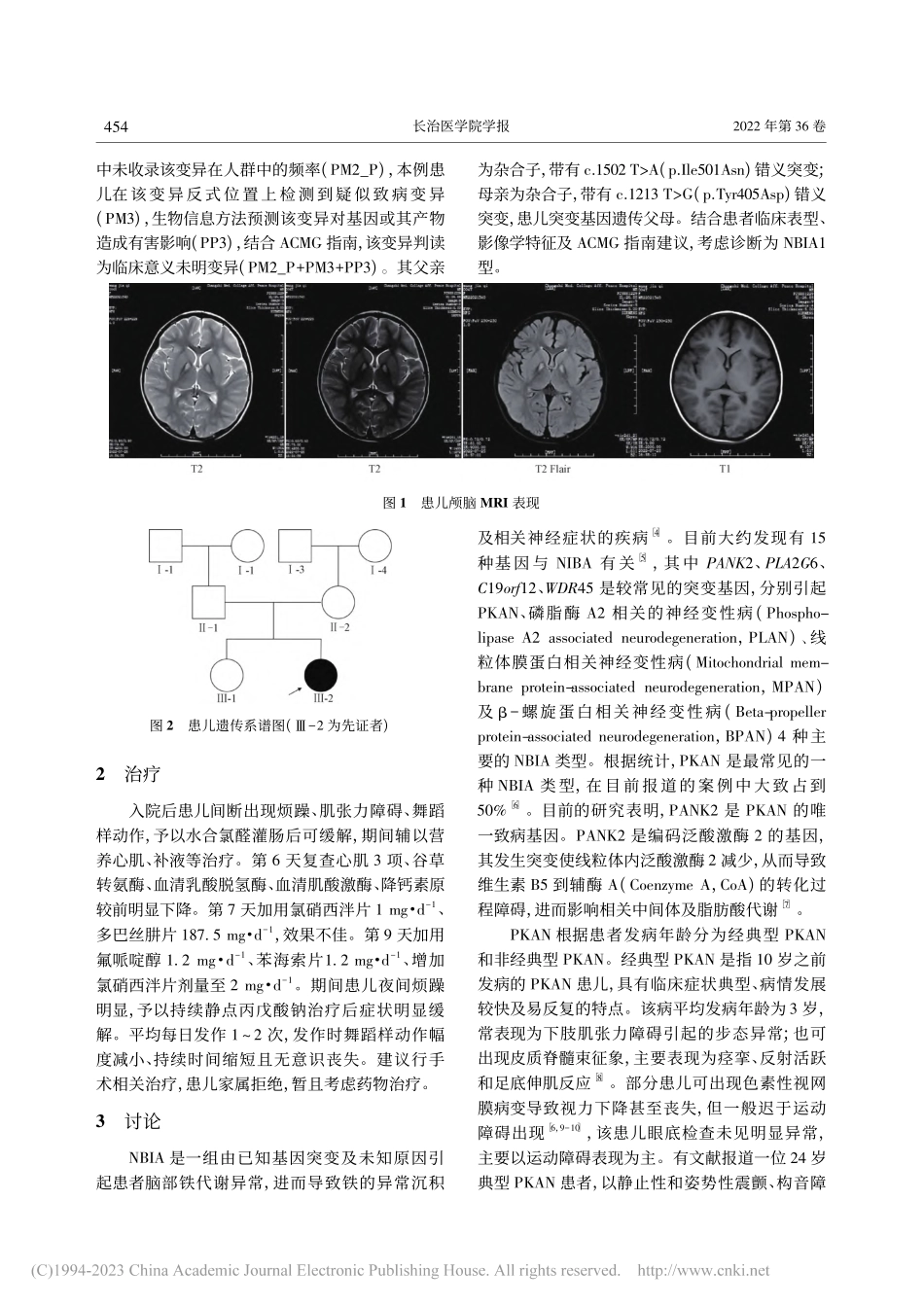
第36卷第6期2022年12月长治医学院学报JOURNALOFCHANGZHIMEDICAICOLLEGEVol.36No.6Dec.2022作者单位:1长治医学院第一临床学院(046000);2山西医科大学公共卫生学院卫生管理学教研室作者简介:林志宏,男,在读硕士,住院医师,研究方向:腰椎间盘突出症患者疾病管理自我效能;通信作者:郑建中,男,博士,主任,教授,研究方向:慢性病管理,E-mail:zjzhong4183@123.com。大脑铁沉积型神经退行性病变1型1例及文献回顾林志宏1杨建1郑建中2关键词大脑铁沉积型神经退行性病变;泛酸激酶相关的神经变性疾病;泛酸激酶2基因中图分类号R745.1文献标识码A文章编号1006-0588(2022)06-453-04大脑铁沉积型神经退行性病变(Neurodegener-ationwithbrainironaccumulation,NBIA)是由致病基因引起的,具有临床及遗传异质性的疾病。其中,大脑铁沉积型神经退行性病变1型(Neurode-generationwithbrainironaccumulation1,NBIA1,OMIM234200),即泛酸激酶相关的神经变性疾病(Pantothenatekinaseassociatedneurodegeneration,PKAN),是一种最常见的NBIA类型[1],部分研究表明PKAN的发病率约为2/100万人[2]。PKAN是位于20p13-p12.3的泛酸激酶2基因(Panto-thenatekinase2,PANK2)突变所致的,发病机制尚不清楚,其遗传方式为常染色体隐性遗传,主要表现为椎体外系功能障碍、肌张力障碍、认知障碍及视网膜病变[3]。PKAN患者的主要病理改变为苍白球局灶性铁沉积,在T2W1上表现为苍白球区中央高信号被周围低信号环绕,又被特征性地称为“虎眼征”。PKAN尚未有技术成熟且疗效明显的治疗手段,临床实践主要以对症治疗减轻神经症状为主,同时延缓疾病进展并预防并发症,因此PKAN的早期识别与诊断尤为重要。本研究通过回顾分析1例PKAN患儿病例,探讨其诊治过程及本病相关研究进展。1病例资料患儿,女性,4岁,曾于1岁6个月时因发育迟缓就诊于外院,其主要表现为运动发育迟缓,只能走平路,不能上台阶,期间接受规律康复治疗。伴语言发育迟缓,至今仍只能说出简单叠词,如“爸爸、妈妈”等。本次主因不能独立行走入院,出现阵发性全身抽搐,表现为舞蹈样动作,每次持续时间约20min,伴意识不清。患儿为G2P2,足月顺产,出生时体质量为3.7kg,否认产时窒息史,父母表型正常,非近亲结婚,否认家族遗传病史。入院后体格检查:体温37.9℃,脉搏120次·min-1,呼吸29次·min-1,血压91/53mmHg(1mmHg=0.133kPa),身高110cm,体质量15kg。精神烦躁,反应迟钝,心脏、肺部及...