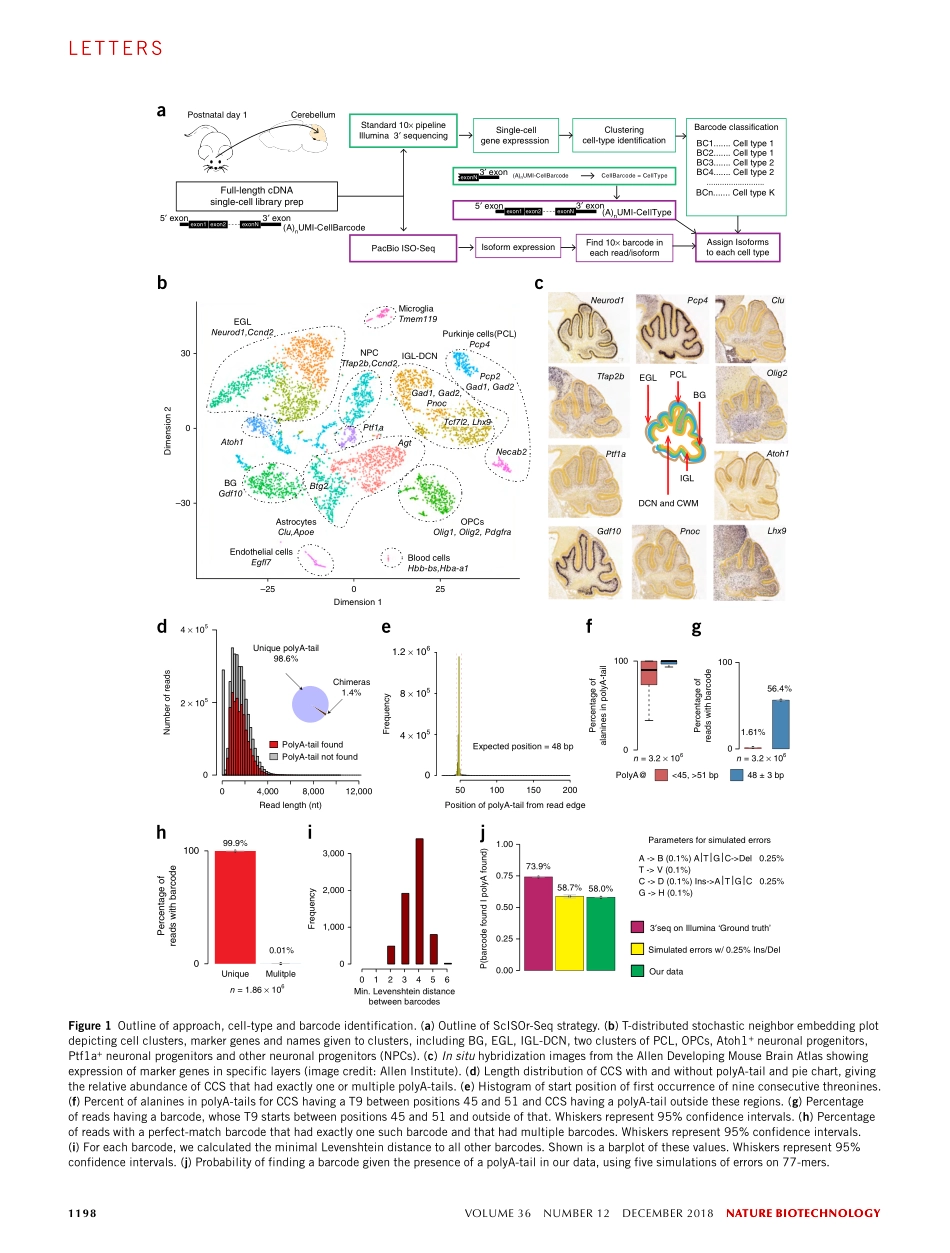
naturebiotechnologyVOLUME36NUMBER12DECEMBER20181197lettersFull-lengthRNAsequencing(RNA-Seq)hasbeenappliedtobulktissue,celllinesandsortedcellstocharacterizetranscriptomes1–11,butapplyingthistechnologytosinglecellshasproventobedifficult,withlessthantensingle-celltranscriptomeshavingbeenanalyzedthusfar12,13.Althoughsinglesplicingeventshavebeendescribedfor≤200singlecellswithstatisticalconfidence14,15,full-lengthmRNAanalysesforhundredsofcellshavenotbeenreported.Single-cellshort-read3′sequencingenablestheidentificationofcellularsubtypes16–21,butfull-lengthmRNAisoformsforthesecelltypescannotbeprofiled.Wedevelopedamethodthatstartswithbulktissueandidentifiessingle-celltypesandtheirfull-lengthRNAisoformswithoutfluorescence-activatedcellsorting.Usingsingle-cellisoformRNA-Seq(ScISOr-Seq),weidentifiedRNAisoformsinneurons,astrocytes,microglia,andcellsubtypessuchasPurkinjeandGranulecells,andcell-type-specificcombinationpatternsofdistantsplicesites6–9,22,23.WeusedScISOr-SeqtoimprovegenomeannotationinmouseGencodeversion10bydeterminingthecell-type-specificexpressionof18,173knownand16,872novelisoforms.Unlikesorting-basedmethods(SupplementaryFig.1a),ScISOr-Seqidentifiesisoformsin>1,000singlecellsfrombulktissuewithoutcellsortingbycombiningtwotechnologies(Fig.1a).Weusedmicrofluid-icstoamplifyfull-lengthcDNAfromsinglecellsinasample.cDNAproducedfromeachsinglecellwasbarcodedtoenablecell-of-originidentificationandthensplitintotwopools,withonepoolbeingusedforshort-readIllumina3′sequencingtomeasuregeneexpressionandtheotherpoolbeingusedforlong-readsequencingandisoformidentification.Short-read3′sequencingprovidedmolecularcountsforeachgeneandcell,whichenabledclusteringofcellsandcelltypeassignmentusingcell-type-specificmarkers.Long-readsequencingwithPacificBiosciences(PacBio)1,2,4,5orOxfordNanopore3wasusedtoidentifyfull-lengthRNAisoforms.Single-cellbarcodeswerealsopresentinlongreadsandcouldbeusedtodeterminetheindividualcelloforiginforeachlongread.Giventhatmostsinglecellsareassignedtoanam...