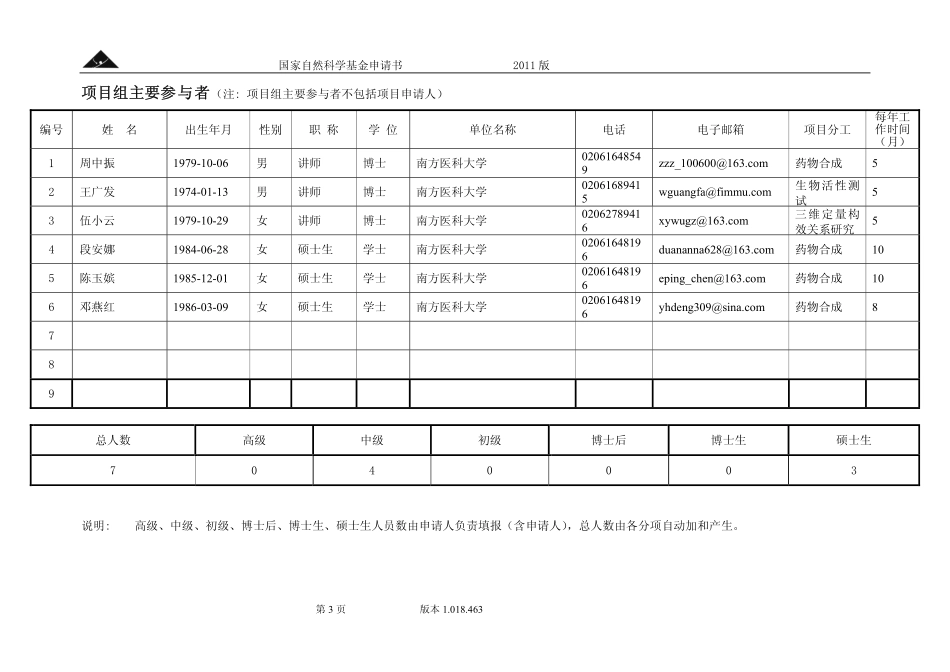
申请代码B020601受理部门收件日期受理编号国家自然科学基金申请书(2011版)资助类别:青年科学基金项目亚类说明:附注说明:项目名称:基于嘧啶核心骨架的新型CDK2抑制剂的设计、合成及生物活性研究申请人:赵培亮电话:02061648196依托单位:南方医科大学通讯地址:广州市沙太南路1023号南方医科大学药学院邮政编码:510515单位电话:020-61648165电子邮箱:peiliangzhao1999@yahoo.com.cn申报日期:2011年3月9日国家自然科学基金委员会国家自然科学基金申请书2011版第2页版本1.018.463基本信息yEqGDFwv姓名赵赵培亮性别男出生年月1980年4月民族汉族学位博士职称讲师每年工作时间(月)10电话02061648196电子邮箱peiliangzhao1999@yahoo.com.cn传真020-61648197国别或地区中国个人通讯地址广州市沙太南路1023号南方医科大学药学院工作单位南方医科大学/药学院申请人信息主要研究领域药物分子的设计、合成及性质研究名称南南方医科大学联系人赵镇电子邮箱nfykdxchengguo@126.com依托单位信息电话020-61648165网站地址www.fimmu.com单位名称合作研究单位信息项目名称基基于嘧啶核心骨架的新型CDK2抑制剂的设计、合成及生物活性研究资助类别青年科学基金项目亚类说明附注说明申请代码B020601:药物分子设计与合成基地类别研究期限2012年1月—2014年12月研究属性基础研究项目基本信息申请经费27.0000万元摘要(限400字):细胞周期是细胞生命活动的基本特征。一个完整的细胞周期受多种蛋白酶的调控,调控失调会导致细胞过度增殖,从而引发肿瘤。以细胞周期蛋白依赖性激酶(cyclin-dependentkinases,CDKs)为靶点的药物可以阻断细胞周期,控制细胞增殖,从而达到抗肿瘤的目的。目前我们组已设计、合成了一批新型CDK2抑制剂,生物活性检测发现化合物YWW-C对肝癌细胞HepG2具有显著的抗肿瘤活性(IC50=9.4μM)。本项目拟在现有工作的基础上,以其为先导化合物,对结构中的A、B、C三个关键部位进行进行结构改造和修饰,并进行活性测试。在此基础上,采用CoMFA、CoMSIA方法从分子的三维结构角度讨论化合物的立体性质、静电性质、疏水性质等与活性之间的关系,建立有较高预测能力的3D-QSAR模型,为化合物结构的优化和修饰提供理论指导,以发展具有进一步修饰与开发潜力的高活性先导化合物。关键词(用分号分开,最多5个)细胞周期蛋白依赖性激酶2;嘧啶骨架;合成;抗肿瘤活性国家自然科学基金申请书2011版第3页版本1.018.463项目组主要参与者(注:项目组主要参与...