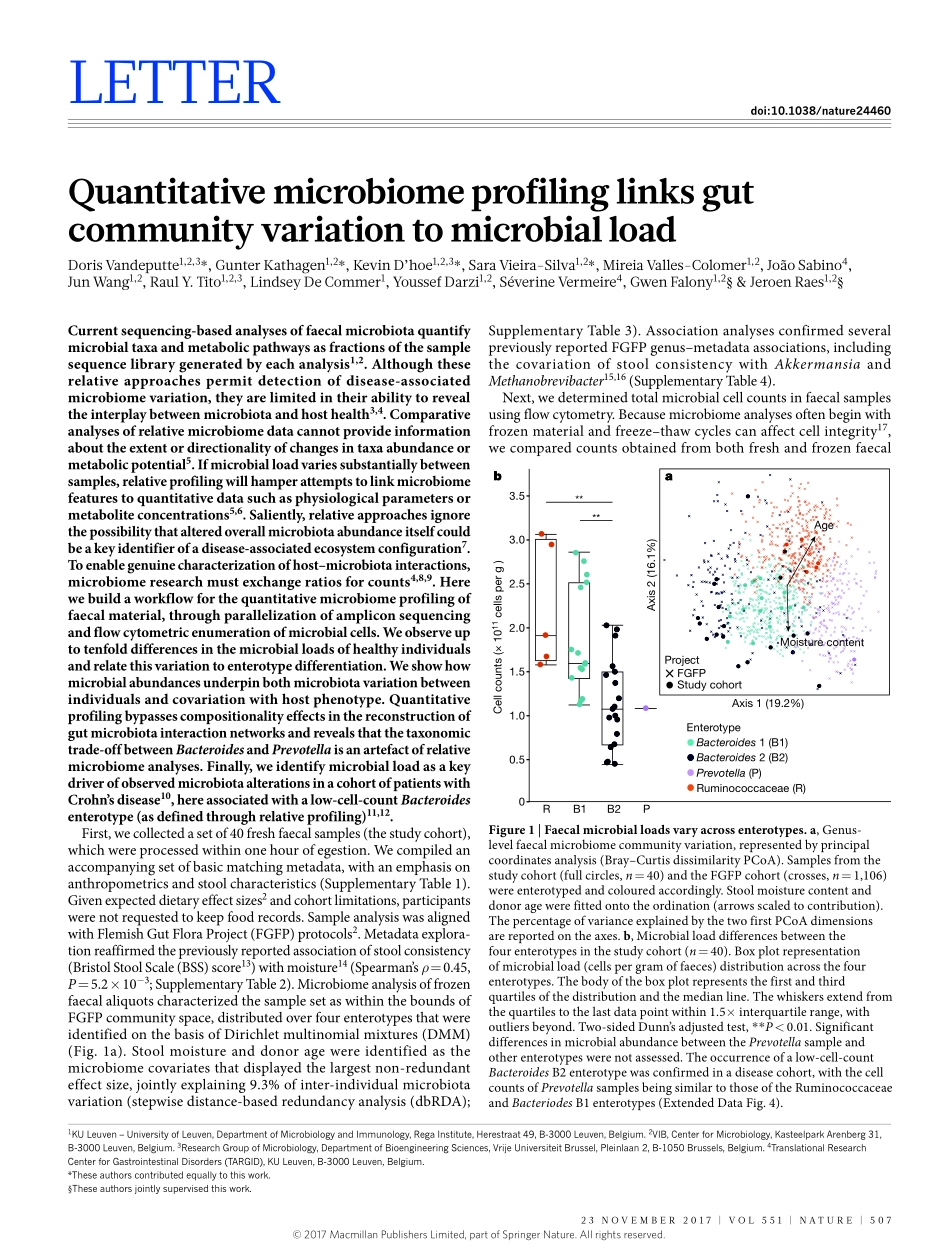
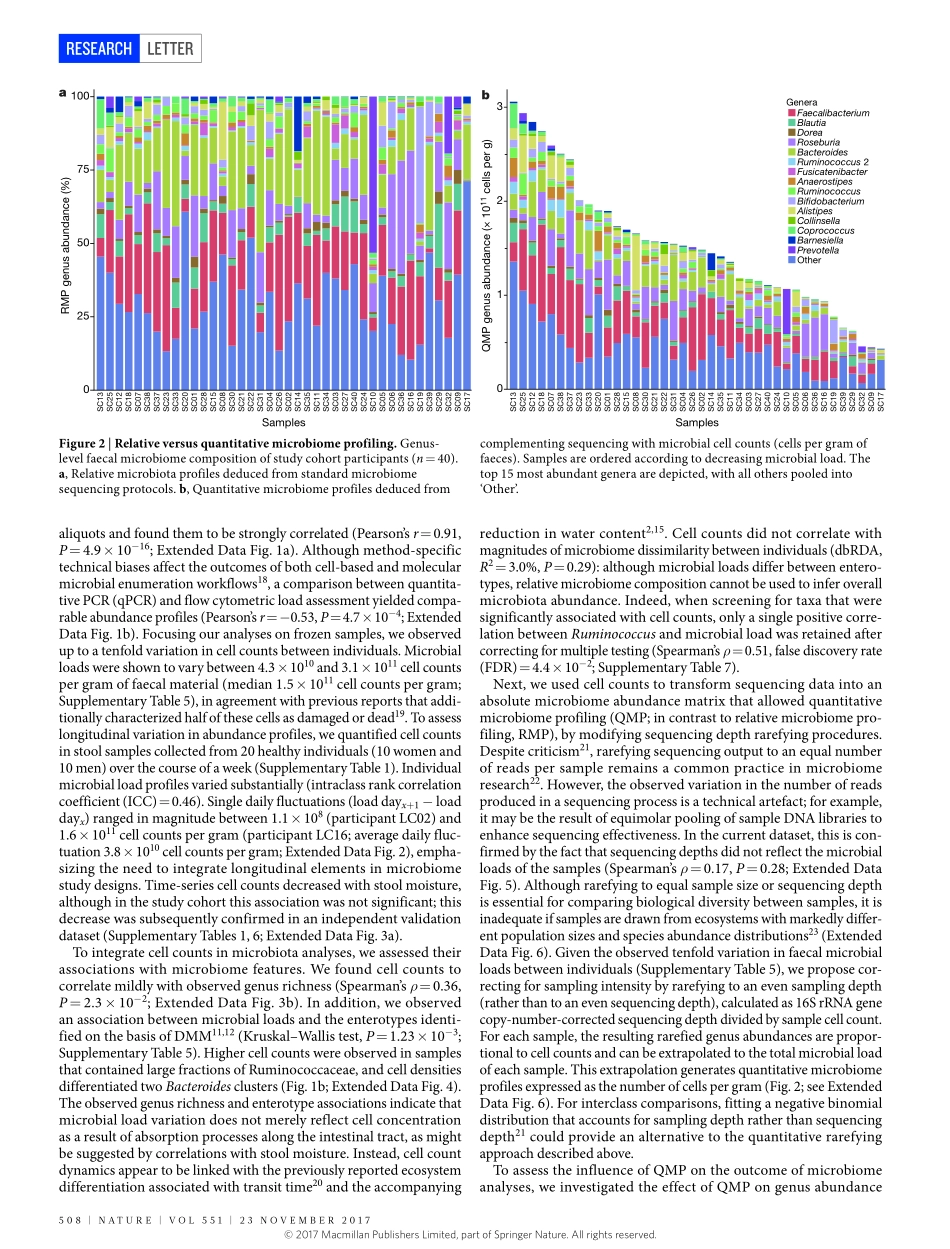
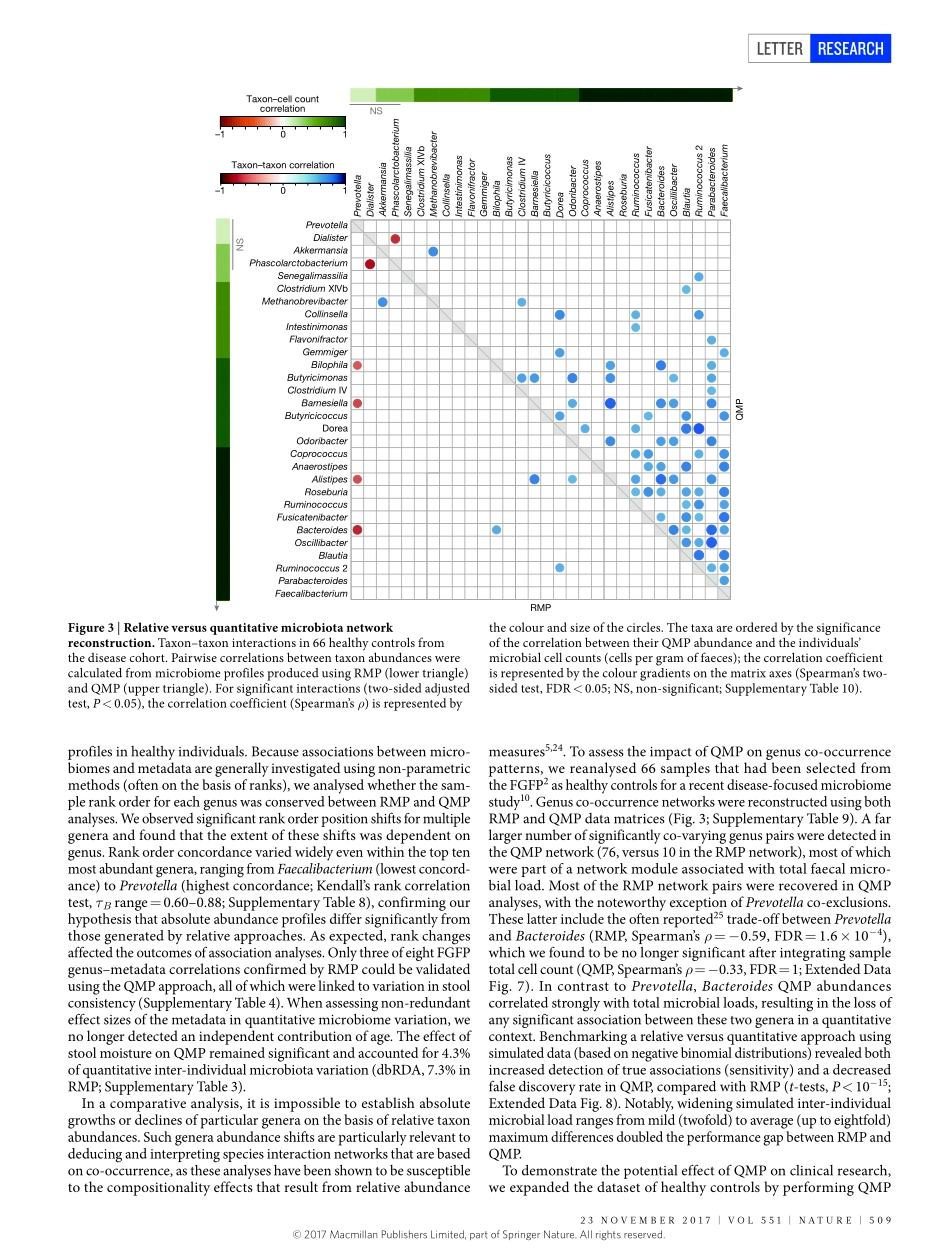
23november2017|voL551|nATUre|507LeTTerdoi:10.1038/nature24460QuantitativemicrobiomeprofilinglinksgutcommunityvariationtomicrobialloadDorisvandeputte1,2,3*,GunterKathagen1,2*,KevinD’hoe1,2,3*,Saravieira-Silva1,2*,mireiavalles-Colomer1,2,JoãoSabino4,JunWang1,2,raulY.Tito1,2,3,LindseyDeCommer1,YoussefDarzi1,2,Séverinevermeire4,GwenFalony1,2§&Jeroenraes1,2§Currentsequencing-basedanalysesoffaecalmicrobiotaquantifymicrobialtaxaandmetabolicpathwaysasfractionsofthesamplesequencelibrarygeneratedbyeachanalysis1,2.Althoughtheserelativeapproachespermitdetectionofdisease-associatedmicrobiomevariation,theyarelimitedintheirabilitytorevealtheinterplaybetweenmicrobiotaandhosthealth3,4.Comparativeanalysesofrelativemicrobiomedatacannotprovideinformationabouttheextentordirectionalityofchangesintaxaabundanceormetabolicpotential5.Ifmicrobialloadvariessubstantiallybetweensamples,relativeprofilingwillhamperattemptstolinkmicrobiomefeaturestoquantitativedatasuchasphysiologicalparametersormetaboliteconcentrations5,6.Saliently,relativeapproachesignorethepossibilitythatalteredoverallmicrobiotaabundanceitselfcouldbeakeyidentifierofadisease-associatedecosystemconfiguration7.Toenablegenuinecharacterizationofhost–microbiotainteractions,microbiomeresearchmustexchangeratiosforcounts4,8,9.Herewebuildaworkflowforthequantitativemicrobiomeprofilingoffaecalmaterial,throughparallelizationofampliconsequencingandflowcytometricenumerationofmicrobialcells.Weobserveuptotenfolddifferencesinthemicrobialloadsofhealthyindividualsandrelatethisvariationtoenterotypedifferentiation.Weshowhowmicrobialabundancesunderpinbothmicrobiotavariationbetweenindividualsandcovariationwithhostphenotype.Quantitativeprofilingbypassescompositionalityeffectsinthereconstructionofgutmicrobiotainteractionnetworksandrevealsthatthetaxonomictrade-offbetweenBacteroidesandPrevotellaisanartefactofrelativemicrobiomeanalyses.Finally,weidentifymicrobialloadasakeydriverofobservedmicrobiotaalterationsinacohortofpatientswithCrohn’sdisease10,...