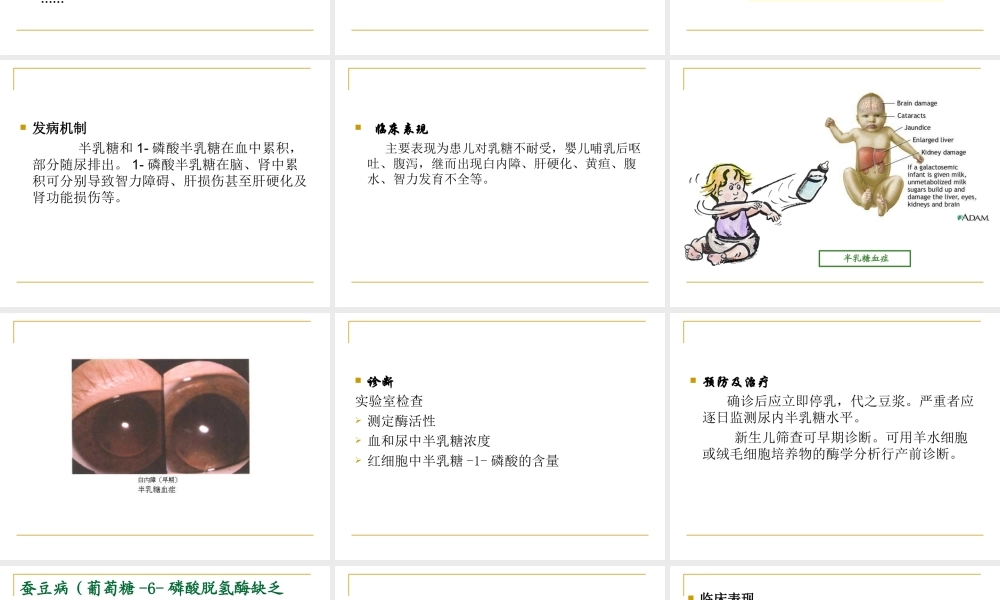
11单基因遗传病(二)Singlegenedisorders单基因遗传病——先天性代谢缺陷前言先天性代谢缺陷的共同规律糖代谢疾病氨基酸代谢疾病核酸代谢疾病前言先天性代谢缺陷(inbornerrorsofmetabolism)基因突变所引起的酶的结构改变或合成障碍,都有可能引起某种代谢过程的中断或紊乱。如果这种基因突变恰好发生在生殖细胞或受精卵中,就有可能传递给后代,从而使后代产生相应的先天性代谢缺陷(或遗传性酶病(hereditaryenzymopathy)。首先提出“先天性代谢缺陷”概念的Garrod先天性代谢缺陷的共同规律酶缺陷与酶活性底物堆积和产物缺乏底物分子的大小与性质临床表型与酶缺陷先天性代谢缺陷疾病的类型糖代谢遗传病氨基酸代谢遗传病核酸代谢遗传病脂类代谢遗传病维生素代谢遗传病金属代谢遗传病药物代谢遗传病……糖代谢障碍半乳糖血症(galactosemia)遗传学半乳糖-1-磷酸尿苷酰转移酶(GPUT)缺陷,本病为常染色体隐性遗传,致病基因定位于9p13,发病率约为1/50000。二、糖代谢病(一)半乳糖血症—具三种亚型:I;II;III半乳糖半乳糖酸半乳糖醇半乳糖-1-磷酸葡萄糖-1-磷酸葡萄糖-6-磷酸半乳糖激酶半乳糖-1-磷酸尿苷转移酶*磷酸葡萄糖变位酶尿苷二磷酸半乳糖-4-表异构酶I型II型III型9p1317q241p35-36半乳糖代谢途径*具有多态性发病机制半乳糖和1-磷酸半乳糖在血中累积,部分随尿排出。1-磷酸半乳糖在脑、肾中累积可分别导致智力障碍、肝损伤甚至肝硬化及肾功能损伤等。临床表现主要表现为患儿对乳糖不耐受,婴儿哺乳后呕吐、腹泻,继而出现白内障、肝硬化、黄疸、腹水、智力发育不全等。半乳糖血症诊断实验室检查测定酶活性血和尿中半乳糖浓度红细胞中半乳糖-1-磷酸的含量预防及治疗确诊后应立即停乳,代之豆浆。严重者应逐日监测尿内半乳糖水平。新生儿筛查可早期诊断。可用羊水细胞或绒毛细胞培养物的酶学分析行产前诊断。蚕豆病(葡萄糖-6-磷酸脱氢酶缺乏症)遗传学X连锁不完全显性,基因定位于Xq28,G6PD缺陷。分布具有世界性,我国主要分布在长江以南,尤其是广东。发病机制G6PD6-p-glucoseGSHG6PD缺陷,导致GSH生成减少,细胞抗氧化损伤能力下降,引起红细胞膜损伤;血红蛋白β链93位半胱氨酸巯基氧化,四聚体解离,形成Heinz小体,红细胞变形能力下降,破坏出现溶血。蚕豆、伯氨喹啉等富含氧化物,服用后可加重该病。临床表现常见于10岁以下小儿,1—5岁为发病高峰。进食蚕豆后,蚕豆中的物质可使患儿...