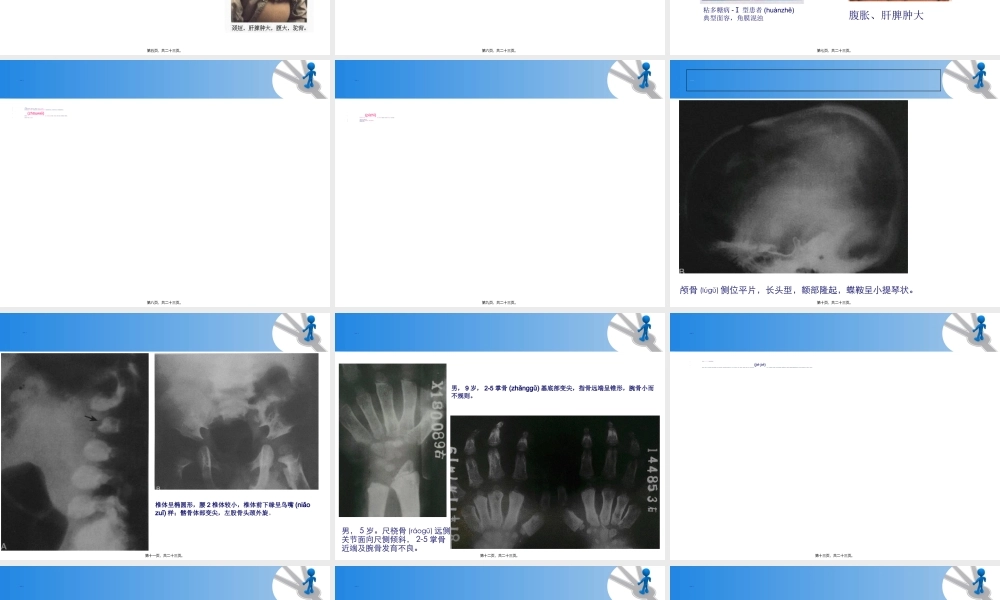
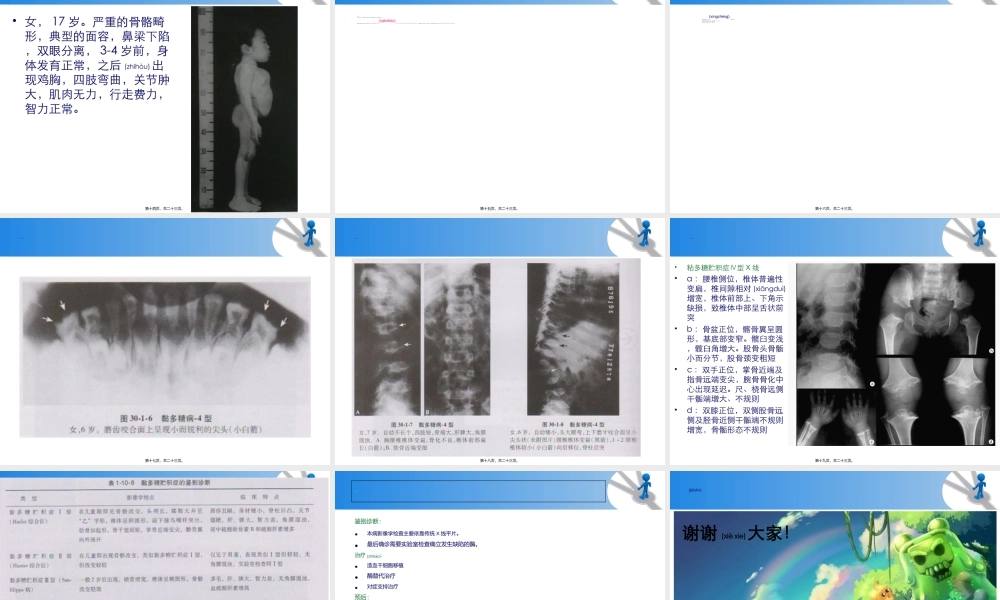
粘多糖贮积(zhùjī)症概述(ɡàishù)及常见临床与X线表现第一页,共二十三页。疾病(jíbìng)的概述•黏多糖贮积症•又名黏多糖代谢障碍(mucopolysaccharidosis,MPS)。•是一种遗传性粘多糖代谢障碍性疾病,造成(zàochénɡ)骨骼、内脏和智力上广泛的失常。第二页,共二十三页。发病的原因(yuányīn)及机制•粘多糖贮积症是由于人体细胞的溶酶体内降解粘多糖的水解酶发生突变导致其活性丧失,粘多糖不能被降解代谢,最终贮积在体内而发生的疾病。•除了Ⅱ型为X链隐性遗传外,其他类型(lèixíng)的粘多糖贮积症均为常染色体隐形遗传,即父母均是该病致病基因的携带者,没有任何粘多糖病的临床症状,但父母同时将该病有异常的基因传给了孩子,使孩子该病对应基因的2条等位基因都出现异常,从而不能生成降解粘多糖的酶。•由于此种遗传病父母双方均为致病基因携带者,故多见于近亲婚配者的子女。第三页,共二十三页。疾病(jíbìng)的分型•该病是溶酶体贮积病中非常重要的一类,根据临床表现、尿中粘多糖的类型及遗传特点等,将MPS分为7型,包括:Ⅰ,Ⅱ,Ⅲ,Ⅳ,Ⅵ,Ⅶ,Ⅸ型等7种型(Ⅴ已归为第Ⅰ型亚型,即MPS-Ⅰ-S),其中Ⅲ又分ⅢA,BⅢ,CⅢ,DⅢ四个亚型,Ⅳ型分为ⅣA和ⅣB亚型。•Ⅰ型和Ⅳ型为该病典型表现。•虽然(suīrán)各型致病基因和临床表现有差异,但由于贮积的底物都是粘多糖而被统称为粘多糖贮积症。•中国人Ⅱ型最常见,其次是ⅣA型。Ⅸ型全世界范围内罕见,目前只有2例报道。第四页,共二十三页。疾病(jíbìng)的诊断当在临床上遇到面容丑陋、骨骼畸形、肝脾肿大、智力低下的患儿时需考虑粘多糖病的可能,确诊需要相关的辅助检查(jiǎnchá):•1.骨骼X线检查:检查部位包括头颅、脊柱、胸部、四肢等•2.尿液粘多糖检测:包括粘多糖定量检测方法和琼脂糖凝胶电泳分析,是粘多糖病的重要筛查方法之一。•3.酶学分析•4.DNA分析第五页,共二十三页。粘多糖病-Ⅰ型•本病亦称Hurler综合征。为染色体隐形遗传。患者多在婴儿或儿童期显示病态,一般预后不良,多在10岁内死亡,常死于呼吸道感染或心力衰竭•临床特点(tèdiǎn):发病常在婴儿期或小儿期,侏儒表现随年龄增大而逐渐显著。患者头大、颈短、肩高、驼背、脊柱侧弯。面容丑陋,鼻梁凹陷,舌大,唇外翻,两眼分离,两耳低下。角膜混浊,耳聋,出齿晚,牙齿不规则,心肌畸形,心力衰竭,肝脾肿大。四肢关节活动受限。智力落后,性功能不良。第六页,共二十三页。粘多...